20 Information Retrieval from Biological Databases
1982
1984
1986
1988
1990
1992
1994
1996
1998
2000
2002
2004
2006
2008
2010
2012
2014
2016
Base pairs
(squares, billions)
Sequences
(circles, millions)
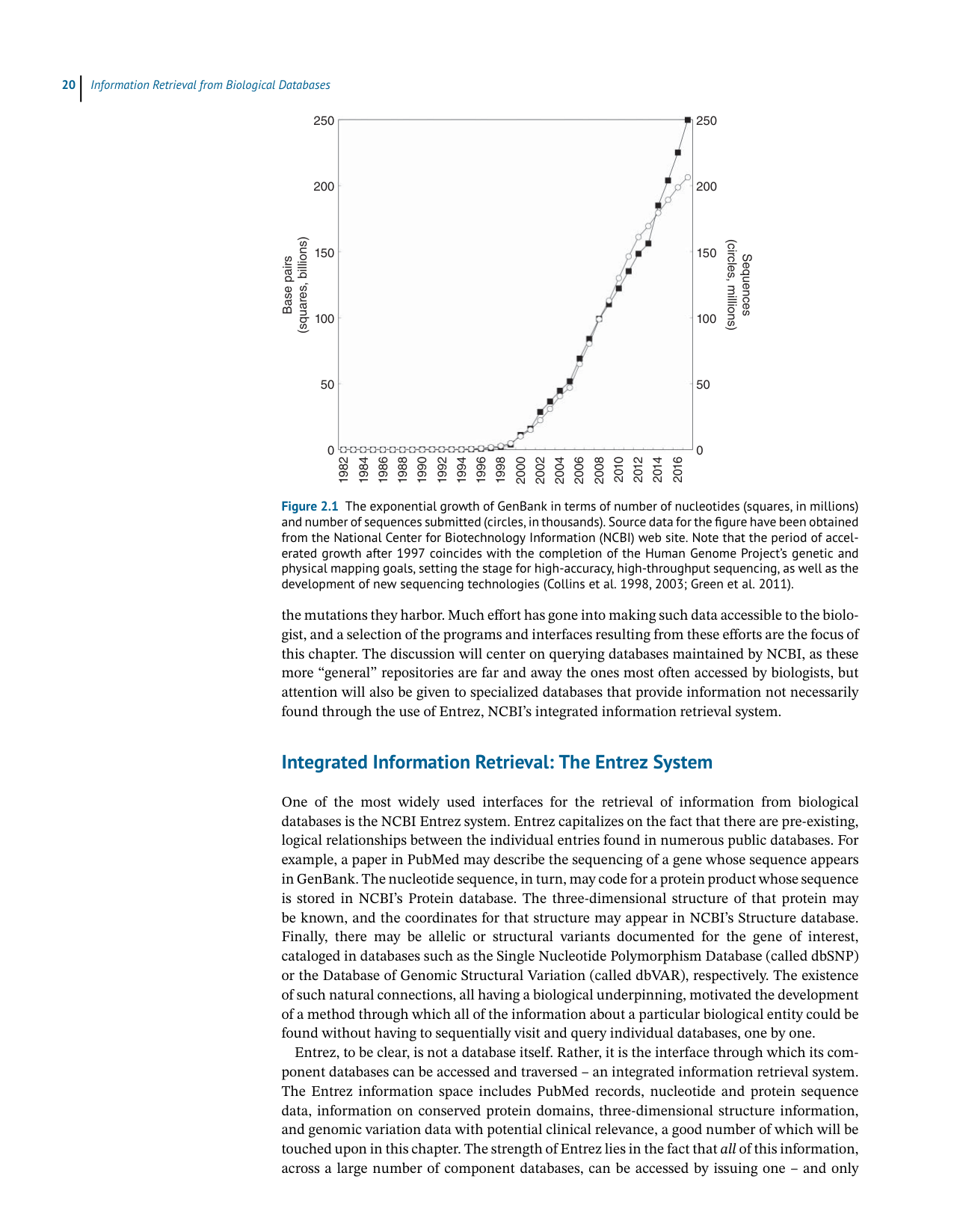
Figure 2.1 The exponential growth of GenBank in terms of number of nucleotides (squares, in millions)
and number of sequences submitted (circles, in thousands). Source data for the figure have been obtained
from the National Center for Biotechnology Information (NCBI) web site. Note that the period of accel-
erated growth after 1997 coincides with the completion of the Human Genome Project’s genetic and
physical mapping goals, setting the stage for high-accuracy, high-throughput sequencing, as well as the
development of new sequencing technologies (Collins et al. 1998, 2003; Green et al. 2011).
themutationstheyharbor.Muchefforthasgoneintomakingsuchdataaccessibletothebiolo-
gist,andaselectionoftheprogramsandinterfacesresultingfromtheseeffortsarethefocusof
thischapter.ThediscussionwillcenteronqueryingdatabasesmaintainedbyNCBI,asthese
more“general”repositoriesarefarandawaytheonesmostoftenaccessedbybiologists,but
attentionwillalsobegiventospecializeddatabasesthatprovideinformationnotnecessarily
foundthroughtheuseofEntrez,NCBI’sintegratedinformationretrievalsystem.
Integrated Information Retrieval: The Entrez System
One of the most widely used interfaces for the retrieval of information from biological
databasesistheNCBIEntrezsystem.Entrezcapitalizesonthefactthattherearepre-existing,
logicalrelationshipsbetweentheindividualentriesfoundinnumerouspublicdatabases.For
example,apaperinPubMedmaydescribethesequencingofagenewhosesequenceappears
inGenBank.Thenucleotidesequence,inturn,maycodeforaproteinproductwhosesequence
is stored in NCBI’s Protein database. The three-dimensional structure of that protein may
be known, and the coordinates for that structure may appear in NCBI’s Structure database.
Finally, there may be allelic or structural variants documented for the gene of interest,
catalogedindatabasessuchastheSingleNucleotidePolymorphismDatabase(calleddbSNP)
ortheDatabaseofGenomicStructuralVariation(calleddbVAR),respectively.Theexistence
ofsuchnaturalconnections,allhavingabiologicalunderpinning,motivatedthedevelopment
ofamethodthroughwhichalloftheinformationaboutaparticularbiologicalentitycouldbe
foundwithouthavingtosequentiallyvisitandqueryindividualdatabases,onebyone.
Entrez,tobeclear,isnotadatabaseitself.Rather,itistheinterfacethroughwhichitscom-
ponentdatabasescanbeaccessedandtraversed–anintegratedinformationretrievalsystem.
The Entrez information space includes PubMed records, nucleotide and protein sequence
data, information on conserved protein domains, three-dimensional structure information,
andgenomicvariationdatawithpotentialclinicalrelevance,agoodnumberofwhichwillbe
toucheduponinthischapter.ThestrengthofEntrezliesinthefactthat allofthisinformation,
across a large number of component databases, can be accessed by issuing one – and only
Integrated Information Retrieval: The Entrez System 21
one–query.Thisverypowerful,integratedapproachismadepossiblethroughtheuseoftwo
generaltypesofconnectionsbetweendatabaseentries: neighboringandhardlinks.
Relationships Between Database Entries: Neighboring
The concept of neighboring enables entrieswithin a given database to be connected to one
another. If a user is lookingat a particular PubMed entry, the user can then “ask” Entrez to
findalloftheotherpapersinPubMedthataresimilarinsubjectmattertotheoriginalpaper.
Likewise,ifauserislookingatasequenceentry,Entrezcanreturnalistofallothersequences
thatbearsimilaritytotheoriginalsequence.Theestablishmentofneighboringrelationships
withinadatabaseisbasedonstatisticalmeasuresofsimilarity,someofwhicharedescribedin
moredetailbelow.Whiletheterm“neighboring”hastraditionallybeenusedtodescribethese
connections,theterminologyontheNCBIwebsitedenotesneighborsas“relateddata.”
BL AST Biologicalsequencesimilaritiesaredetectedandsequencedataarecomparedwithone
another using the Basic Local Alignment Search Tool, or BLAST (Altschul et al. 1990). This
algorithmattemptstofindhigh-scoringsegmentpairs–pairsofsequencesthatcanbealigned
with one another and, when aligned, meet certain scoring and statistical criteria. Chapter 3
discussesthefamilyofBLASTalgorithmsandtheirapplicationatlength.
VAST Molecular structure similarities are detected and sets of coordinate data are com-
pared using a vector-based method known as VAST (the Vector Alignment Search Tool;
Gibrat et al. 1996). This methodology uses geometric criteria to assess similarity between
three-dimensionaldomains,andtherearethreemajorstepsthattakeplaceinthecourseofa
VASTcomparison:
• First,basedon knownthree-dimensionalcoordinatedata,thealphahelicesandbetastrands
thatconstitutethestructuralcoreofeachproteinareidentified.Straight-linevectorsarethen
calculatedbasedonthepositionofthesesecondarystructuralelements.VASTkeepstrack
of how one vector is connected to the next (that is, how the C-terminal end of one vector
connects to the N-terminal end of the next vector), as well as whether each vector repre-
sents an alphahelix or a beta strand. Subsequent comparison steps useonlythese vectors
inassessingstructuralsimilaritytootherproteins–so,ineffect,mostofthepainstakingly
deducedatomiccoordinatedataarediscardedatthisstep.Thereasonforthisapparentover-
simplificationissimplyduetothescaleoftheproblemathand;withthe150000structures
intheMolecularModelingDatabase(MMDB;Madejetal.2014)availableatthetimeofthis
writing,thetimethatitwouldtaketodoanin-depthcomparisonofeachandeveryoneof
thesestructureswithalloftheotherstructuresinMMDBwouldmakethecalculationsboth
impracticalandintractable.
• Next, the algorithm attempts to optimally align these sets of vectors, looking for pairs of
structuralelementsthatareofthesametypeandrelativeorientation,withconsistentcon-
nectivity between the individual elements. The object is to identify highly similar “core
substructures,”pairsthatrepresentastatisticallysignificantmatchabovethatwhichwould
beobtainedbycomparingrandomlychosenproteinswithoneanother.
• Finally,arefinementisdoneusingMonteCarlo(randomsearch)methodsateachresidue
positiontooptimizethestructuralalignment.Theresultantalignmentneednotbeglobal,
asmatchesmaybebetweenindividualstructuraldomainsoftheproteinsbeingcompared.
In 2014, a significant improvement to VAST was introduced. This new approach, called
VAST+ (Madej et al. 2014), moves beyond assessing structural similarity by comparing
individual three-dimensional domains with one another; instead, it considers the entire set
of three-dimensional domains within a macromolecular complex. This approach essentially
movesthecomparisonfromthetertiarystructuretothequaternarystructurelevel,enabling
the identification of similar functional, multi-subunit assemblies. In the VAST+ parlance,
macromolecularcomplexesarereferredtoasa“biologicalunit”andcanincludenotjustthe
22 Information Retrieval from Biological Databases
proteinsthatconstitutethecomplex,butalsonucleotidesandchemicalswheresuchstructural
informationisavailable.TheVAST +comparisonbeginsasdescribedaboveforVASTandthen
marchesthroughanumberofstepsthatinvolvetheidentificationofbiologicalunitsthatcan
be superimposed, calculation of root-mean-square deviations (RMSDs) of the superimposed
structuresasaquantitativemeasureofthesuperposition(seeBox12.1),and,finally,performs
arefinementsteptoimprovetheRMSDvaluesforthesuperposition.Theresultofthisprocess
is a global structural alignment where both the most and least similar parts of the aligned
moleculescanbeidentifiedand,fromabiologicalstandpoint,comparisonsbetweensimilarly
shapedproteinscanbefacilitated;itcanalsobeusedinthecontextoflookingatconforma-
tionalchangesofasinglecomplexundervaryingconditions.WhileVAST +isnowthedefault
methodforidentifyingstructuralneighborswithintheEntrezsystem,keepinmindthatthe
algorithm depends on biological units being explicitly identified within the source Protein
Data Bank (PDB) coordinate data records that form the basis for MMDB records; if no such
biologicalunitsaredefined,theoriginalVASTalgorithmisthenusedforthecomparisons.
ByusingapproachessuchasVASTandVAST +,itispossibletofindstructuralrelationships
betweenproteinsincaseswheresimplylookingatsequencesimilaritymaynotsuggestrelat-
edness–informationthatcould,withadditionaldataandinsights,beusedtohelpinformthe
questionoffunctionalsimilarity.Moreinformationonadditionalstructurepredictionmeth-
ods based on X-ray or nuclear magnetic resonance (NMR) coordinate data can be found in
Chapter12.
Weighted Key T erms The problemofcomparingsequence or structure data somewhatpales
nexttothatofcomparingPubMedentries,whichconsistoffreetextwhoserulesofsyntaxare
notnecessarilyfixed.Giventhatnotwopeople’swritingstylesareexactlythesame,findinga
waytocompareseeminglydisparateblocksoftextposesasubstantialproblem.Entrezemploys
amethodknownastherelevancepairsmodelofretrievaltomakesuchcomparisons,relying
onweightedkeyterms(WilburandCoffee1994;WilburandYang1996).Thisconceptisbest
describedbyexample.Considertwomanuscriptswiththefollowingtitles:
BRCA1asaGeneticMarkerforBreastCancer
GeneticFactorsintheFamilialTransmissionoftheBreastCancerBRCA1Gene
Bothtitlescontaintheterms BRCA1,Breast,and Cancer,andthepresenceofthesecommon
termsmayindicatethatthemanuscriptsaresimilarinsubjectmatter.Theproximitybetween
thewordsisalsoconsidered,sothatwordscommontotworecordsthatareclosertogetherare
scoredhigherthancommonwordsthatarefurtherapart.Intheexample,theterms Breastand
Cancerarealwaysnexttoeachother,sotheywouldscorehigherbasedonproximitythaneither
ofthosewordswouldagainst BRCA1.Commonwordsfoundinatitlescorehigherthanthose
foundinanabstract,sincetitlewordsarepresumedtobe“moreimportant”thanthosefound
in the body of an abstract. Overall, weighting depends inversely on the frequency of a given
word among all the entries in PubMed, with words that occur infrequently in the database
assignedahigherweightwhilecommonwordsaredown-weighted.
Hard Links
The hard link concept is simpler and much more straightforward than the neighboring
approachesdescribedabove.Hardlinksareappliedbetweenentriesin differentdatabasesand
existwhereverthereisalogicalconnectionbetweenentries.Forinstance,ifaPubMedentry
describesthesequencingofachromosomalregioncontainingageneofinterest,ahardlinkis
establishedbetweenthePubMedentryandthecorrespondingnucleotideentryforthatgene.
If an open reading frame in that gene codes for a known protein, a hard link is established
betweenthenucleotideentryandtheproteinentry.Iftheproteinentryhasanexperimentally
deducedstructure,ahardlinkwouldbeplacedbetweentheproteinentryandthestructural
entry.
SearchescanbeginanywherewithintheEntrezecosystem–therearenoconstraintsonthe
user as to where the foray into this information space must begin. However, depending on
whichdatabaseisusedasthejumping-offpoint,differentdatabasefieldswillbeavailablefor
Integrated Information Retrieval: The Entrez System 23
searching.Thisstandstoreason,astheentriesindifferentdatabasesarenecessarilyorganized
differently,reflectingthebiologicalnatureoftheentitiesthateachdatabaseistryingtocatalog.
The Entrez Discovery Pathway
The best way to illustrate the integrated nature of the Entrez system and to drive home the
powerofneighboringisbyconsideringsomebiologicalexamples.Thesimplestwaytoquery
Entrezisthroughtheuseofindividualsearchterms,coupledtogetherbyBooleanoperators
such as AND, OR, or NOT. Consider the case in which one wants to retrieve all available
information on a gene namedDCC (deleted in colorectal carcinoma), limiting the returned
informationtopublicationswhereaninvestigatornamedGuyA.Rouleauisanauthor.There
isaverysimplequeryinterfaceatthetopoftheNCBIhomepage,allowingtheusertoselect
whichdatabasetheywanttosearchfromapull-downmenuandatextboxwherethequery
termscanbeentered.Inthiscase,tosearchforpublishedpapers,PubMedwouldbeselected
fromthepull-downmenuand,withinthetextboxtotheright,theuserwouldtype DCC AND
"Rouleau GA" [AU] .The [AU]qualifyingthesecondsearchtermindicatestoEntrezthat
this is anauthor term, so only the author field in entries should be considered when evalu-
atingthispartofthesearchstatement.TheresultofthequeryisshowninFigure2.2.Here,
Figure 2.2 Results of a text-based Entrez query against PubMed using Boolean operators and field delimiters. The initial query ( DCC AND
"Rouleau GA" [AU] ) is shown in the search box near the top of the window, with the three papers identified using this query following
below. Each entry gives the title of the manuscript, the names of the authors, and the citation information. The actual record can be
retrieved by clicking on the name of the manuscript.
24 Information Retrieval from Biological Databases
T able 2.1 Entrez Boolean search statements.
General syntax:
search term [tag] Boolean operator search term [tag] ...
where [tag] =
[ACCN] Accession
[AD] Affiliation
[ALL] Allfields
[AU] Authorname
Lentz R [AU] yieldsallof LentzRA,LentzRB,etc.
"Lentz R" [AU] yieldsonly LentzR
[AUID] Uniqueauthoridentifier,suchasanORCIDID
[ECNO] EnzymeCommissionnumbers
[EDAT] Entrezdate
YYYY/MM/DD, YYYY/MM,or YYYY;insertacolonfordaterange,
e.g. 2016:2018
[GENE] Genename
[ISS] Issueofjournal
[JOUR] Journaltitle,officialabbreviation,orISSNnumber
Journal of Biological Chemistry
J Biol Chem
0021-9258
[LA] Language
[MAJR] MeSHmajortopic
Oneofthe majortopicsdiscussedinthearticle
[MH] MeSHterms
Controlledvocabularyofbiomedicalterms( subject)
[ORGN] Organism
[PDAT] Publicationdate
YYYY/MM/DD, YYYY/MM,or YYYY;insertacolonfordaterange,
e.g. 2016:2018
[PMID] PubMedID
[PROT] Proteinname(forsequencerecords)
[PT] Publicationtype,includes:
Review
Clinical Trial
Lectures
Letter
Technical Report
[SH] MeSHsubheading
UsedtomodifyMeSHTerms
stenosis [MH] AND pharmacology [SH]
[SUBS] Substancename
Nameofchemicaldiscussedinarticle
[SI] SecondarysourceID
Namesofsecondarysourcedatabanksand/oraccessionnumbersof
sequencesdiscussedinarticle
[TITL] Titleword
Onlywordsinthedefinitionline(notavailableinStructuredatabase)
[WORD] Text words
Allwordsandnumbersinthetitleandabstract,MeSHterms,
subheadings,chemicalsubstancenames,personalnameassubject,
andMEDLINEsecondarysources
[VOL] Volumeofjournal
andBooleanoperator =
AND, OR,or NOT
Integrated Information Retrieval: The Entrez System 25
threeentriesmatchingthequerywerefoundinPubMed.Theusercanfurthernarrowdown
thequerybyaddingadditionaltermsiftheuserisinterestedinamorespecificaspectofthis
geneoriftherearequitesimplytoomanyentriesreturnedbytheinitialquery.Alistofavailable
fielddelimitersisgiveninTable2.1.
ForeachofthefoundpapersshownintheSummaryviewinFigure2.2,theuserispresented
with the title of the paper, the authors of that paper, and the citation. To look at any of the
papers resulting from the search, the user can simply click on any of the hyperlinked titles.
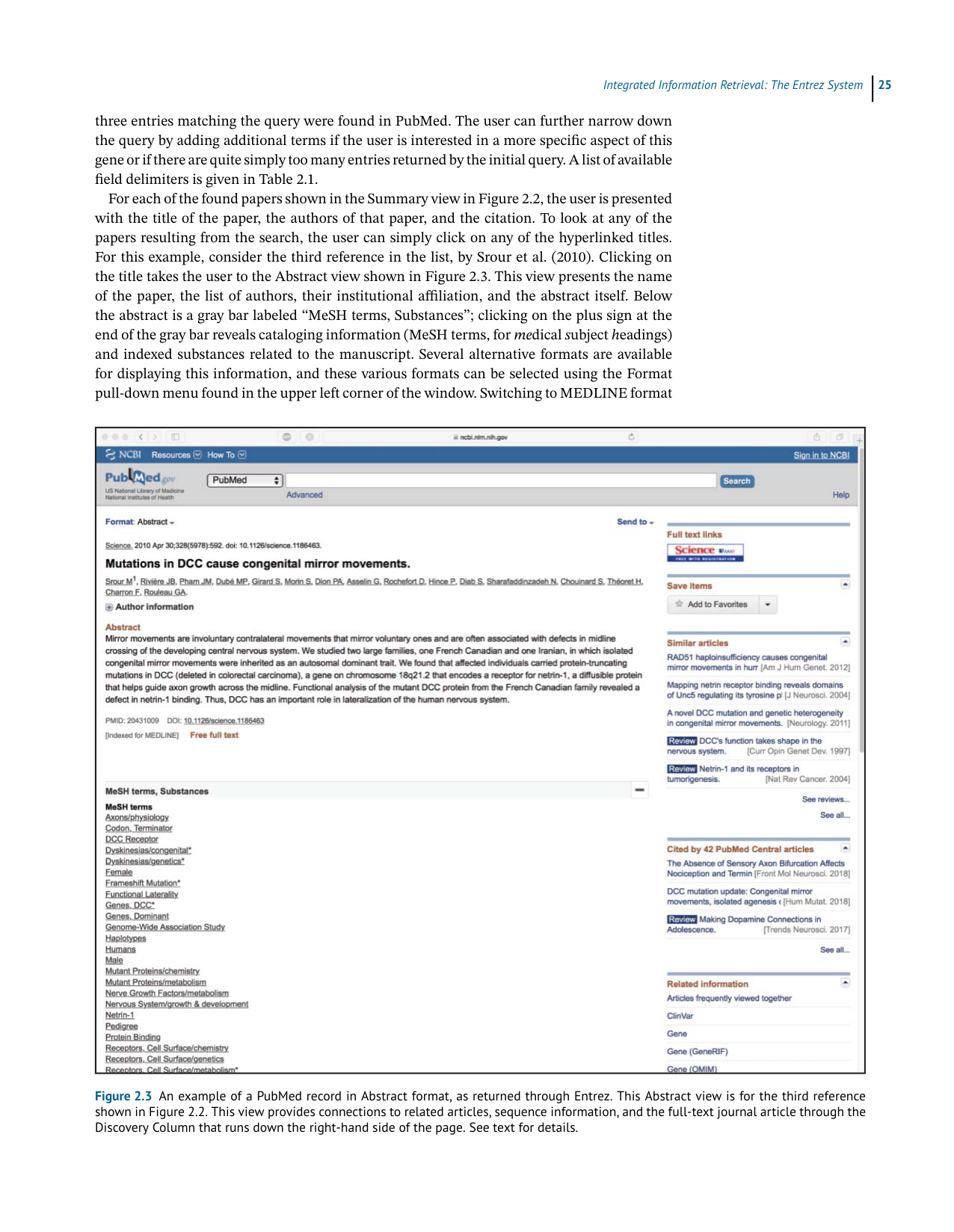
For this example, consider the third reference in the list, by Srour et al. (2010). Clicking on
thetitletakestheusertotheAbstractviewshowninFigure2.3.Thisviewpresentsthename
of the paper, the list of authors, their institutional affiliation, and the abstract itself. Below
theabstractisagraybarlabeled“MeSHterms,Substances”;clickingontheplussignatthe
endofthegraybarrevealscataloginginformation(MeSHterms,for medicalsubjectheadings)
and indexed substances related to the manuscript. Several alternative formats are available
for displaying this information, and these various formats can be selected using the Format
pull-downmenufoundintheupperleftcornerofthewindow.SwitchingtoMEDLINEformat
Figure 2.3 An example of a PubMed record in Abstract format, as returned through Entrez. This Abstract view is for the third reference
shown in Figure 2.2. This view provides connections to related articles, sequence information, and the full-text journal article through the
Discovery Column that runs down the right-hand side of the page. See text for details.
26 Information Retrieval from Biological Databases
produces the MEDLINE layout, with two-letter codes corresponding to the contents of each
fieldgoingdowntheleft-handsideoftheentry(e.g.theauthorfieldisagaindenotedbythe
code AU).Listsofentriesinthisformatcanbesavedtothedesktopandeasilyimportedinto
third-partybibliographymanagementprograms.
The column on the right-hand side of this window – aptly named the Discovery
Column – provides access to the full-text version of the paper and, more importantly,
containsmanyusefullinkstoadditionalinformationrelatedtothismanuscript.TheSimilar
articlessectionprovidesoneoftheentrypointsfromwhichtheusercantakeadvantageofthe
neighboring and hard link relationships described earlier and, in the examples that follow,
we will return to this page several times to illustrate a selected cross-section of the kinds of
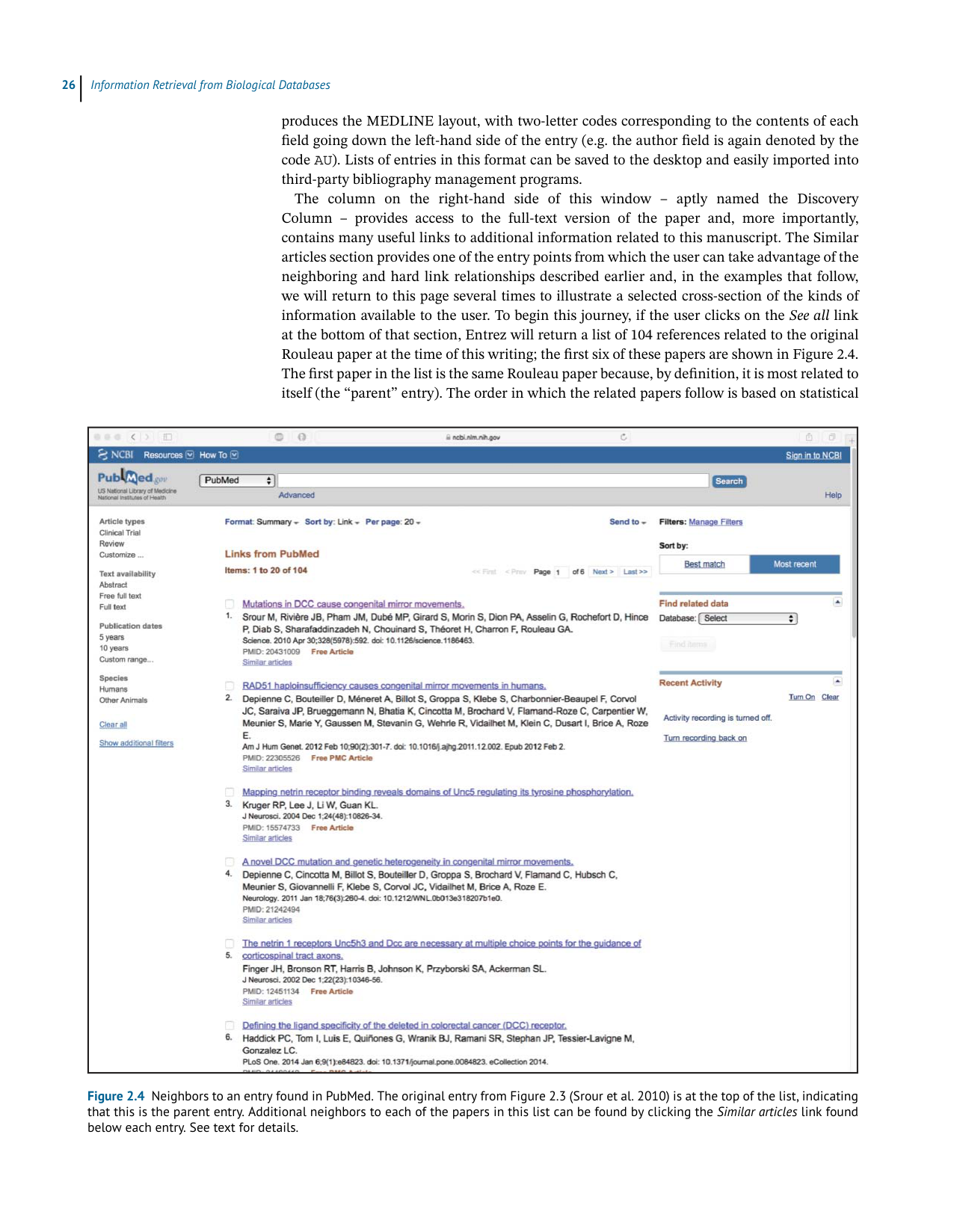
information available to the user. To begin this journey, if the user clicks on theSeeall link
atthebottomofthatsection,Entrezwillreturnalistof104referencesrelatedtotheoriginal
Rouleaupaperatthetimeofthiswriting;thefirstsixofthesepapersareshowninFigure2.4.
ThefirstpaperinthelististhesameRouleaupaperbecause,bydefinition,itismostrelatedto
itself(the“parent”entry).Theorderinwhichtherelatedpapersfollowisbasedonstatistical
Figure 2.4 Neighbors to an entry found in PubMed. The original entry from Figure 2.3 (Srour et al. 2010) is at the top of the list, indicating
that this is the parent entry. Additional neighbors to each of the papers in this list can be found by clicking the Similar articles link found
below each entry. See text for details.
Integrated Information Retrieval: The Entrez System 27
Figure 2.5 The Entrez Gene page for the DCC (deleted in colorectal carcinoma) netrin-1 receptor from human. The entry indicates that this
is a protein-coding gene at map location 18q21.2, and information on the genomic context of DCC, as well as alternative gene names and
information on the encoded protein, is provided. An extensive collection of links to other National Center for Biotechnology Information
(NCBI) and external databases is also provided. See text for details.
similarity. Thus, the entry closest to the parent is deemed to be the closest in subject matter
to the parent. By scanning the titles, the user can easily find related information on other
studies, as well as quickly amass a bibliography of relevant references. This is a particularly
usefulandtime-savingfunctionwhenoneiswritinggrantsorpapers,asabstractscaneasily
bescannedandpapersofrealinterestcanbeidentifiedquickly.
Returning to the Abstract view presented in Figure 2.3, at the bottom of the Discovery
Column is a series of hard-link connections to other databases within the Entrez system
thatcantaketheuserdirectlytoanextensivesetofinformationrelatedtothecontentofthe
publicationofinterest.Here,selectingtheGenelinktakestheusertoEntrezGene,afeatureof
Entrezthatprovidesawealthofinformationaboutthegeneinquestion(Figure2.5).Thedata
aregatheredfromavarietyofsources,includingRefSeq.Here,weseethatDCCistheofficial
symbol of a protein-coding gene for a netrin-1 receptor in humans. The Genomic context
sectionofthispageindicatesthattheDCCisaprotein-codinggeneatmaplocation18q21.2.
28 Information Retrieval from Biological Databases
Immediately below, summary information on the genomic region, transcripts, and products
of theDCC gene are presented graphically, with genomic coordinates provided. Additional
content not shown in the figure can be found by scrolling down the Gene page, where the
user will find relevant functional information (such as gene expression data), associated
phenotypes,informationonprotein–proteininteractions,pathwayinformation,GeneOntol-
ogyassignments,andhomologiestosimilarsequencesinselectedorganisms.Shortcutlinksto
thesesectionscanbefoundintheTableofcontentsatthetopoftheDiscoveryColumn.Fur-
therdowntheDiscoveryColumnareextensivelistsoflinkstoadditionalresourcesprovided
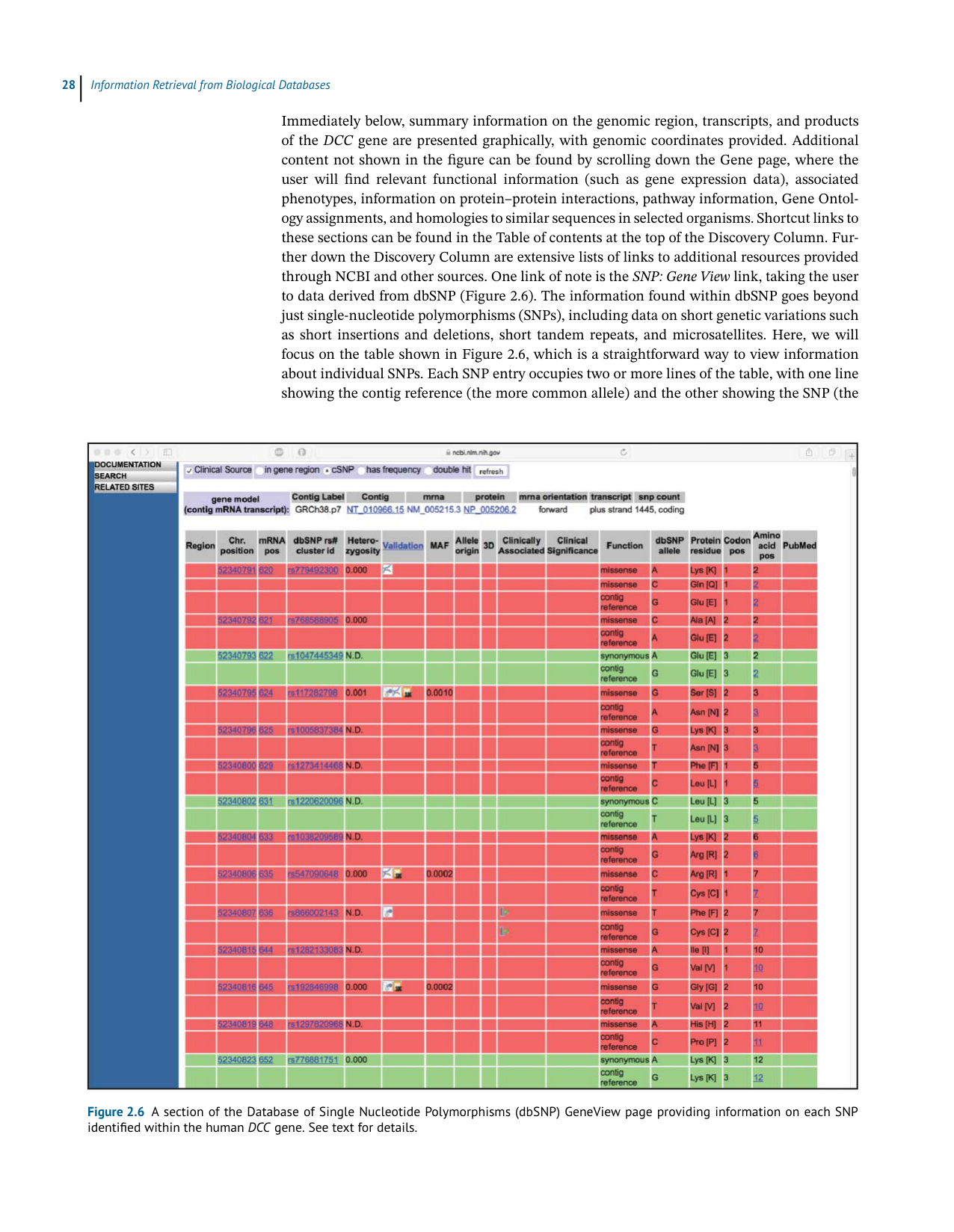
throughNCBIandothersources.Onelinkofnoteisthe SNP:GeneView link,takingtheuser
todataderivedfromdbSNP(Figure2.6).TheinformationfoundwithindbSNPgoesbeyond
justsingle-nucleotidepolymorphisms(SNPs),includingdataonshortgeneticvariationssuch
as short insertions and deletions, short tandem repeats, and microsatellites. Here, we will
focus on the table shown in Figure 2.6, which is a straightforward way to view information
aboutindividualSNPs.EachSNPentryoccupiestwoormorelinesofthetable,withoneline
showingthecontigreference(themorecommonallele)andtheothershowingtheSNP(the
Figure 2.6 A section of the Database of Single Nucleotide Polymorphisms (dbSNP) GeneView page providing information on each SNP
identified within the human DCC gene. See text for details.
Integrated Information Retrieval: The Entrez System 29
lesscommonallele).Considerthefirstthreelinesofthetable,showingacontigreferenceGfor
whichtherearetwodocumentedSNPs,changingtheGatthatpositiontoeitheranAoraC.At
theproteinlevel,thischangestheaminoacidatposition2oftheDCCproteinfromglutamic
acidtolysine(fortheG-to-Asubstitution)ortoglutamine(fortheG-to-Csubstitution).These
rowsarecoloredredsincetheseare“non-synonymousSNPs”–thatis,theSNPproducesadis-
cretechangeattheaminoacidlevel.Incontrast,considerthefirstsetofgreenrowsinthetable,
with the green indicating that this is a “synonymous SNP,” where the codons for the contig
reference(G)andtheSNPallele(A)ultimatelyproducethesameaminoacid(Glu);thisisnot
altogethersurprising,withtheSNPbeinginthewobblepositionofthecodon,wherethereis
oftenredundancyinthegeneticcode.AdditionalinformationonhumanSNPscanbefoundin
Chapter15.
StartingagainfromtheAbstractviewshowninFigure2.3,proteinsequencesfromRefSeq
that have been linked to this abstract can be found by clicking on theProtein (RefSeq)link
found in the Related information section on the right-hand side of the page, producing the
viewshowninFigure2.7.Notethatallbutoneoftheentriesismarkedas“predicted”;thefinal
Figure 2.7 Entries in the RefSeq protein database corresponding to the original Srour et al. (2010) entry in Figure 2.3. Entries can be
accessed and examined by clicking on any of the accession numbers. See text for details.
30 Information Retrieval from Biological Databases
Figure 2.8 The RefSeq entry for the netrin receptor, the protein product of the human DCC gene. The FASTA link at the top of the entry
provides quick access to the protein sequence in FASTA format, while the Graphics link provides access to a graphical view of all of the
individual elements captured within the entry’s feature table (see Figure 2.9). See text for details.
entry in the list has an accession number beginning with NP, indicating that it contains an
experimentallydeterminedorverifiedsequence(seeBox1.2).Clickingonthefirstlineofthat
entry(number6)takestheusertotheviewshowninFigure2.8,theRefSeqentryforthenetrin
receptor,theproteinproductofthe DCCgene.Thefeaturetable–thesectionoftheGenBank
entry listing the location and characteristics of each of the documented biological features
found within this protein sequence, such as post-translational modifications, recognizable
repeat units, secondary structural regions, and clinically relevant variation – is particularly
long in this case. This makes it difficult to determine the relative orientation of the features
tooneanotherandmayleadtheusertomissimportantinteractionsorrelationshipsbetween
biologicalfeatures.Fortunately,aviewerthatprovidesabird’seyeviewoftheelementsfound
within the feature table is available by clicking on the Graphics link at the top of the entry,
producingthemoreaccessibledisplayshowninFigure2.9.Zoomcontrolsareprovided,and
Integrated Information Retrieval: The Entrez System 31
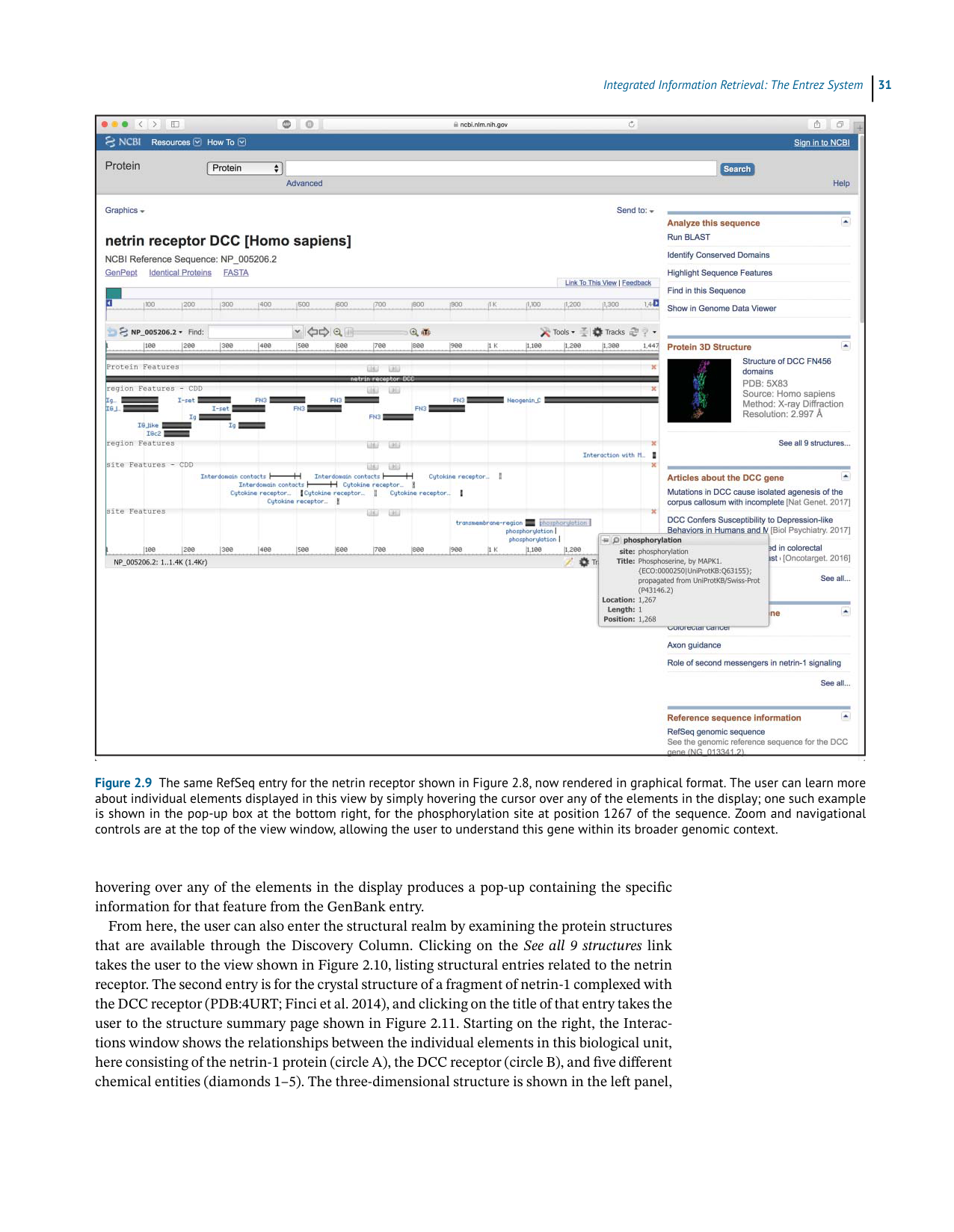
Figure 2.9 The same RefSeq entry for the netrin receptor shown in Figure 2.8, now rendered in graphical format. The user can learn more
about individual elements displayed in this view by simply hovering the cursor over any of the elements in the display; one such example
is shown in the pop-up box at the bottom right, for the phosphorylation site at position 1267 of the sequence. Zoom and navigational
controls are at the top of the view window, allowing the user to understand this gene within its broader genomic context.
hovering over any of the elements in the display produces a pop-up containing the specific
informationforthatfeaturefromtheGenBankentry.
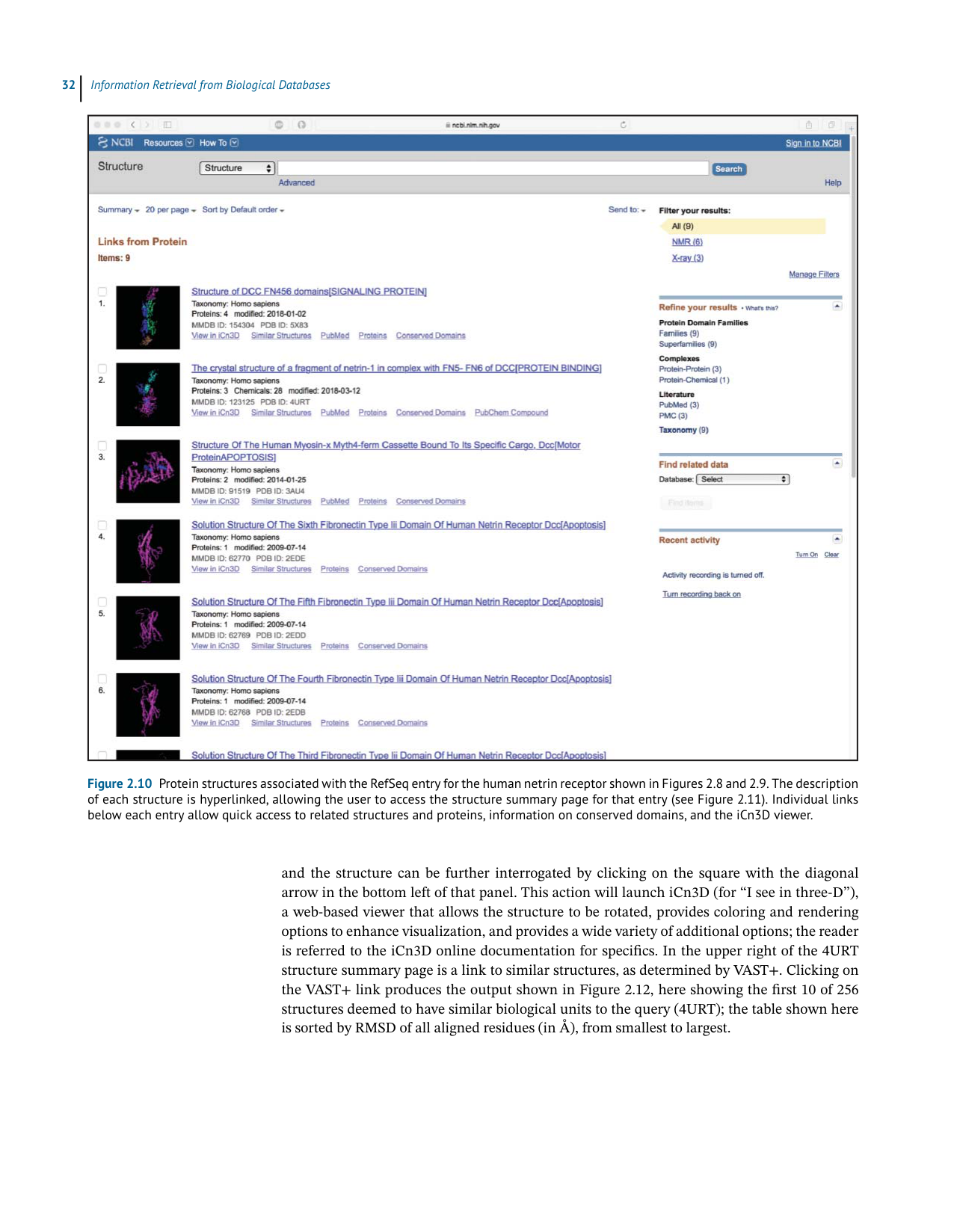
Fromhere,theusercanalsoenterthestructuralrealmbyexaminingtheproteinstructures
that are available through the Discovery Column. Clicking on theSee all 9 structureslink
takestheusertotheviewshowninFigure2.10,listingstructuralentriesrelatedtothenetrin
receptor.Thesecondentryisforthecrystalstructureofafragmentofnetrin-1complexedwith
theDCCreceptor(PDB:4URT;Fincietal.2014),andclickingonthetitleofthatentrytakesthe
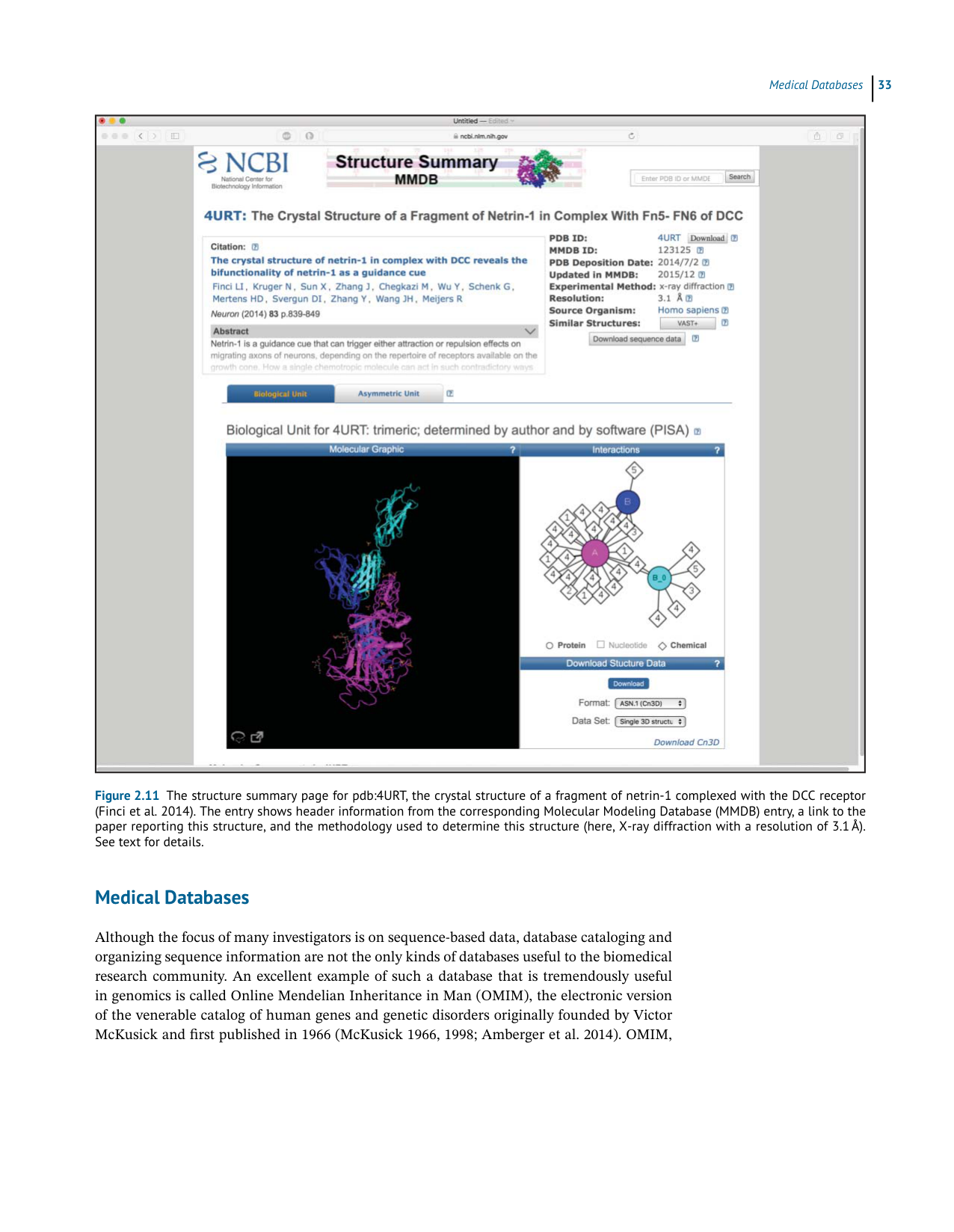
usertothestructuresummarypageshowninFigure2.11.Startingontheright,theInterac-
tionswindowshowstherelationshipsbetweentheindividualelementsinthisbiologicalunit,
hereconsistingofthenetrin-1protein(circleA),theDCCreceptor(circleB),andfivedifferent
chemicalentities(diamonds1–5).Thethree-dimensionalstructureisshownintheleftpanel,
32 Information Retrieval from Biological Databases
Figure 2.10 Protein structures associated with the RefSeq entry for the human netrin receptor shown in Figures 2.8 and 2.9. The description
of each structure is hyperlinked, allowing the user to access the structure summary page for that entry (see Figure 2.11). Individual links
below each entry allow quick access to related structures and proteins, information on conserved domains, and the iCn3D viewer.
and the structure can be further interrogated by clicking on the square with the diagonal
arrowinthebottomleftofthatpanel.ThisactionwilllaunchiCn3D(for“Iseeinthree-D”),
a web-based viewer that allows the structure to be rotated, provides coloring and rendering
optionstoenhancevisualization,andprovidesawidevarietyofadditionaloptions;thereader
is referred to the iCn3D online documentation for specifics. In the upper right of the 4URT
structuresummarypageisalinktosimilarstructures,asdeterminedbyVAST +.Clickingon
the VAST+link produces the output shown in Figure 2.12, here showing the first 10 of 256
structuresdeemedtohavesimilarbiologicalunitstothequery(4URT);thetableshownhere
issortedbyRMSDofallalignedresidues(inÅ),fromsmallesttolargest.
===== PDF page 53 (Figure 2.11 caption portion) =====
Medical Databases 33
Figure 2.11 The structure summary page for pdb:4URT , the crystal structure of a fragment of netrin-1 complexed with the DCC receptor
(Finci et al. 2014). The entry shows header information from the corresponding Molecular Modeling Database (MMDB) entry, a link to the
paper reporting this structure, and the methodology used to determine this structure (here, X-ray diffraction with a resolution of 3.1 Å).
S e et e x tf o rd e t a i l s .
Text-Specific Search Options: Limits and Advanced Search
实际小节名:Medical Databases
用途:
Medical Databases 33
Figure 2.11 The structure summary page for pdb:4URT , the crystal structure of a fragment of netrin-1 complexed with the DCC receptor
(Finci et al. 2014). The entry shows header information from the corresponding Molecular Modeling Database (MMDB) entry, a link to the
paper reporting this structure, and the methodology used to determine this structure (here, X-ray diffraction with a resolution of 3.1 Å).
S e et e x tf o rd e t a i l s .
Medical Databases
Althoughthefocusofmanyinvestigatorsisonsequence-baseddata,databasecatalogingand
organizingsequenceinformationarenottheonlykindsofdatabasesusefultothebiomedical
research community. An excellent example of such a database that is tremendously useful
in genomicsis calledOnlineMendelianInheritancein Man(OMIM), the electronicversion
of the venerable catalog of human genes and genetic disorders originally founded by Victor
McKusick and first published in 1966 (McKusick 1966, 1998; Amberger et al. 2014). OMIM,
34 Information Retrieval from Biological Databases
Figure 2.12 A list of structures deemed similar to pdb:4URT using VAST +. The table is sorted by the root-mean-square deviation of all
aligned residues (in Å), from smallest to largest. Details on each individual structure in the list can be found by clicking on its Protein Data
Bank (PDB) ID number.
which is authored and maintained at The Johns Hopkins University School of Medicine,
providesconcisetextualinformationfromthepublishedliteratureonmosthumanconditions
havingageneticbasis,aswellaspicturesillustratingtheconditionordisorder(whereappro-
priate), full citation information, and links to a number of useful external resources, some
of which will be described below. As will become obvious through the following example, a
basicknowledgeofOMIMshouldbepartofthearmamentariumofphysician-scientistswith
aninterestintheclinicalaspectsofgeneticdisorders.
OMIMhasadefinednumberingsysteminwhicheachentryisassignedauniquenumber–a
“MIM number” – that is similar to an accession number, with certain positions within that
number indicating information about the genetic disorder itself. The first digit represents
the mode of inheritance of the disorder: 1, 2, and 6 stand for autosomal loci or phenotypes,
3 for X-linked loci or phenotype, 4 for Y-linked loci or phenotype, and 5 for mitochondrial
lociorphenotypes.Anasterisk(*)precedingaMIMnumberindicatesagene,ahashsign(#)
indicates an entry describing a phenotype, a plus sign (+) indicates that the entry describes
Medical Databases 35
Figure 2.13 Online Mendelian Inheritance in Man (OMIM) entries related to the DCC gene. The hash sign (#) preceding the first entry
indicates that it is an entry describing a phenotype – here, mirror movements. The second entry is preceded by an asterisk (*), indicating
that it is a gene entry – here, for the DCC gene.
a gene of known sequence and phenotype, and a percent sign (%) describes a confirmed
Mendelian phenotype or locus for which the underlying molecular basis is unknown. If no
Mendelian basis has been clearly established for a particular entry, no symbol precedes the
MIMnumber.
Here,wewillcontinuetheEntrezexamplefromtheprevioussection,followingthe OMIM
(cited)linkfoundintheDiscoveryColumnshowninFigure2.3.Anintermediatelandingpage
will then appear listing two entries, one for theDCC gene, the other for a phenotype entry
describingmirrormovements(Figure2.13).Clickingonthesecondentryleadstheusertothe
OMIMpageforthe DCCgeneshowninFigure2.14,withtheTextsectionoftheentryproviding
acomprehensiveoverviewofseminaldetailsregardingtheidentificationofthegene,itsstruc-
ture,relevantbiochemicalfeatures,mappinginformation,anoverviewofthegene’sfunction
andmoleculargenetics,andstudiesinvolvinganimalmodels.Forindividualsstartingworkon
anewgeneorgeneticdisorder,thisexpertlycuratedsectionoftheOMIMentryshouldbecon-
sidered“requiredreading,”asitpresentsthemostimportantaspectsofanygivengene,with
36 Information Retrieval from Biological Databases
Figure 2.14 The Online Mendelian Inheritance in Man (OMIM) entry for the DCC gene. Each entry in OMIM includes information such as
the gene symbol, alternative names for the disease, a description of the disease, a clinical synopsis, and references. See text for details.
links to the original studies cited within the narrative embedded throughout. A particularly
usefulfeatureisthelistofallelicvariants(Figure2.15);ashortdescriptionisgivenaftereach
allelicvariantoftheclinicalorbiochemicaloutcomeofthatparticularmutation.Atthetime
of this writing, there are over 5200 OMIM entries containing at least one allelicvariant that
eithercausesorisassociatedwithadiscretephenotypeinhumans.Notethattheallelicvari-
ants shown in Figure 2.15 produce significantly different clinical outcomes – two different
types of cancer as well as the motor disorder used throughout this example – an interesting
casewheredifferentmutationsinthesamegeneleadtodistinctgeneticdisorders.
The studies leading to these and similar observations described in a typical entry often
provide the foundation for clinical trials aimed at translating this knowledge into new
preventionandtreatmentstrategies.NIH’scentralinformationsourceforclinicaltrials,aptly
named ClinicalTrials.gov, contains data on both publicly and privately funded clinical trials
Medical Databases 37
Figure 2.15 An example of a list of allelic variants that can be found through Online Mendelian Inheritance in Man (OMIM). The figure
shows three of the four allelic variants for the DCC gene. T wo of the documented variants lead to cancers of the digestive tract, while
two are associated with a movement disorder. The description under each allelic variant provides information specific to that particular
mutation.
beingconductedworldwide.Figure2.16showsthefirsteightofmorethan4600clinicaltrials
actively recruiting patients with colorectal cancer at the time of this writing, and clicking
on the name of a protocol will bring the user to a page providing information on the study,
includingtheprincipalinvestigator’snameandcontactinformation.Clickingthe OnMap tab
atthetopofthepageproducesaclickablemapoftheworldshowinghowmanyclinicaltrials
arebeingconductedineachregionorcountry(Figure2.17);thisviewisusefulinidentifying
trialsthataregeographicallyclosetoapotentialstudysubject’shome.Whilewe,asscientists,
tend to focus on the types of information discussed throughout the rest of this chapter, the
clinical trials site is, unarguably, the most important of the sites covered in this chapter, as
it provides a means through which patients with a given genetic or metabolic disorder can
38 Information Retrieval from Biological Databases
Figure 2.16 The ClinicalT rials.gov page showing all actively recruiting clinical trials relating to colorectal neoplasms. Information on each
trial, including the principal investigator of the trial and qualification criteria for participating in the trial, can be found by clicking on the
name of the trial.
receivethelatest,cutting-edgetreatment–treatmentthatmaymakeasubstantialdifference
totheirqualityoflife.
Organismal Sequence Databases Beyond NCBI
Although it may appear from this discussion that NCBI is the center of the sequence uni-
verse,manyspecializedgenomicdatabasesthroughouttheworldservespecificgroupsinthe
scientificcommunity.Often,thesedatabasesprovideadditionalinformationnotavailableelse-
where,suchasphenotypes,experimentalconditions,straincrosses,andmapfeatures.These
dataareofgreatimportancetothesecommunities,notonlybecausetheyinfluenceexperimen-
tal design and interpretation of experimental results but also because the kinds of data they
containdonotalwaysfitneatlywithintheconfinesoftheNCBIdatamodel.Developmentof
specialized databases necessarily ensued (and continues), and these databases are intended
tobeusedasanimportantadjuncttoGenBankandsimilarglobaldatabases.Itisimpossible